Capítulo 23 Regressão filogenética
Esse capítulo implementa contrastes filogenéticos independentes (PIC) (Felsenstein 1985), regressão filogenética generalizada por quadrados mínimos (PGLS) (Grafen 1989) e regressão de autovetores filogenéticos (PVR) (Diniz-Filho, de Sant’Ana, and Bini 1998).
23.1 Contrastes filogenéticos independentes
Vamos investigar a relação evolutiva entre força da mordida e tamanho do crânio em um gênero de roedores. Pra isso, vamos precisar dos dados fenotípicos e pelo menos uma hipótese filogenética.
Carregar dados fenotípicos.
dados<-read.table("dadospcm/akodon.txt",h=T)
dados
attach(dados)Antes, vamos ajustar um modelo ordinário (OLS), sem levar a filogenia em consideração, apenas como exemplo didático. Na prática, você não deve/precisa fazer isso.
# OLS
fit.ols<-lm(bite_force~skull_length,data=dados)
summary(fit.ols)
{plot(skull_length,bite_force,
xlab="skull length (log)",
ylab="bite force (log)",pch=21,bg="grey",cex=1.5)
abline(fit.ols,col="red")}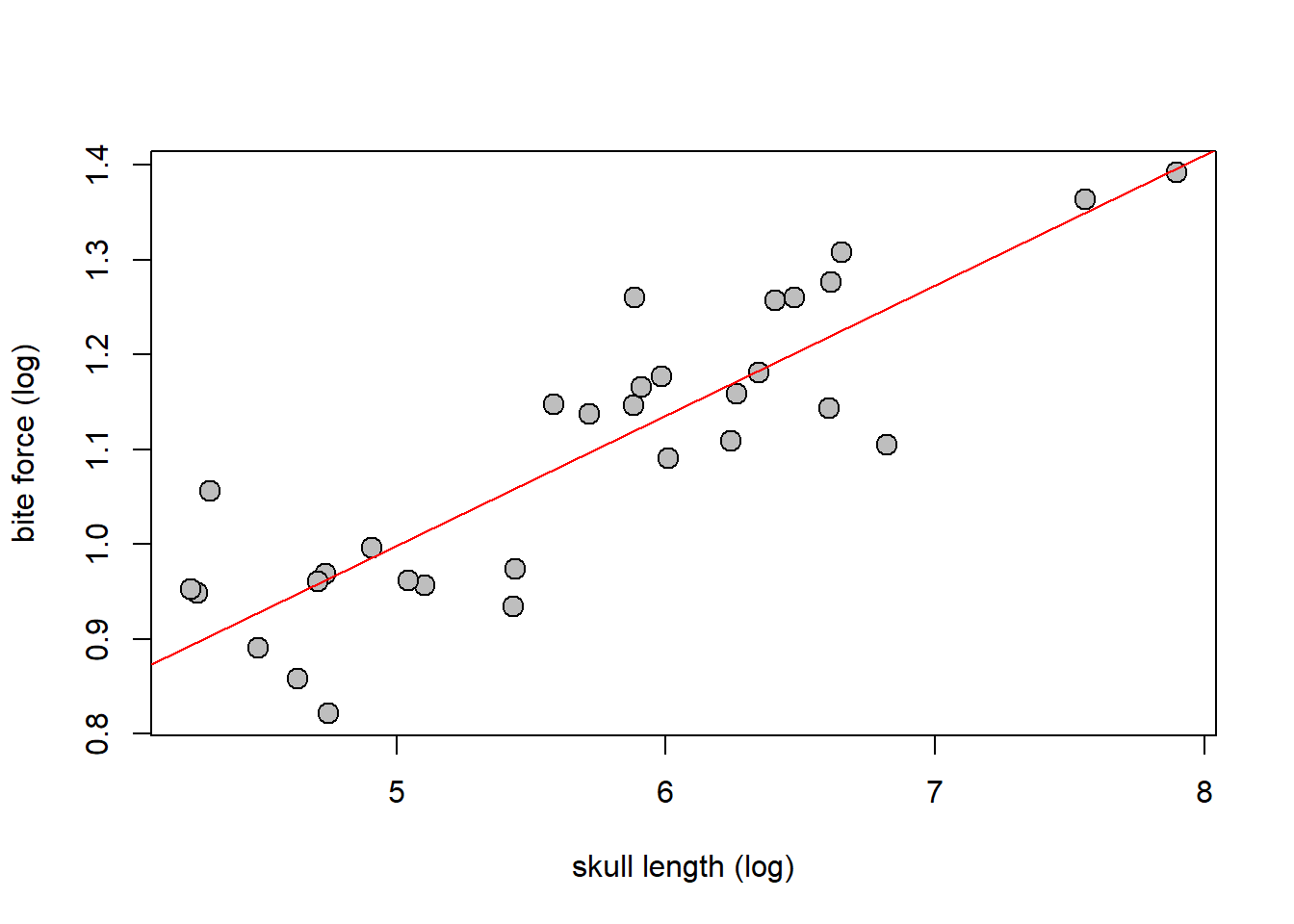
Como os dados são de espécies diferentes, o pressuposto de independência dos resíduos do modelo é violado. Uma maneira de levar em consideração a filogenia é através dos Contrastes Filogenéticos Independentes.
# PIC
require(ape)
#> Le chargement a nécessité le package : ape
akodon.tree<-read.tree(file="dadospcm/akodon.tree")
plot(akodon.tree)
akodon.tree
# Atribuindo nomes para os dados
skull_length<-setNames(skull_length,species)
skull_length
bite_force<-setNames(bite_force,species)
bite_force
# Calculando os contrastes
pic.sl<-pic(skull_length,akodon.tree)
pic.bf<-pic(bite_force,akodon.tree)
# Modelo PIC
fit.pic<-lm(pic.bf~pic.sl+0)
fit.pic
summary(fit.pic)
plot(pic.sl,pic.bf,
xlab="PICs for skull length",
ylab="PICs for bite force",pch=21,bg="grey",cex=1.5)
abline(fit.pic,col="red")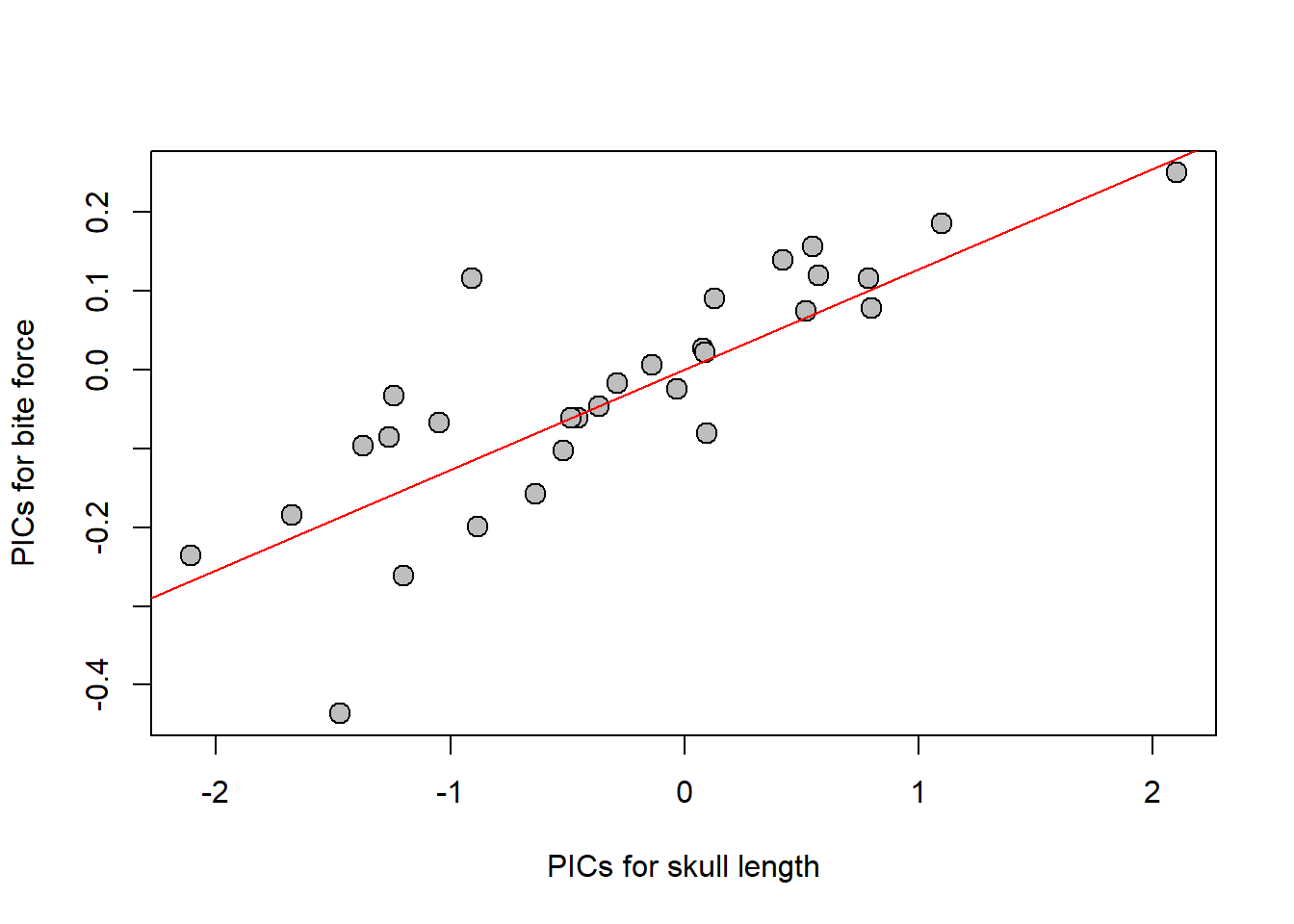
Como podemos perceber, para estes dados não existe muita diferença nos resultados levando em consideração a filogenia. Isso nem sempre vai ser o caso. Nós podemos simular condições para visualizar como levar em conta a filogenia pode ter uma consequência grande.
# PIC dados simulados
require(phytools)
#> Le chargement a nécessité le package : phytools
#> Le chargement a nécessité le package : maps
# set seed (starting point na geração de sequência aleatória, assim todos teremos os mesmos resultados)
set.seed(4)
# simular uma árvore filogenética
tree<-rcoal(n=80)
plotTree(tree,ftype="off")
# simular 2 atributos (independentemente) evoluindo por movimento Browniano na filogenia
x<-fastBM(tree)
y<-fastBM(tree)
par(mar=c(5,4,2,2))
# Ajustar OLS
plot(x,y,pch=21,bg="grey",cex=1.5)
fit.OLS<-lm(y~x)
{plot(x,y,pch=21,bg="grey",cex=1.5)
abline(fit.OLS,col="red")}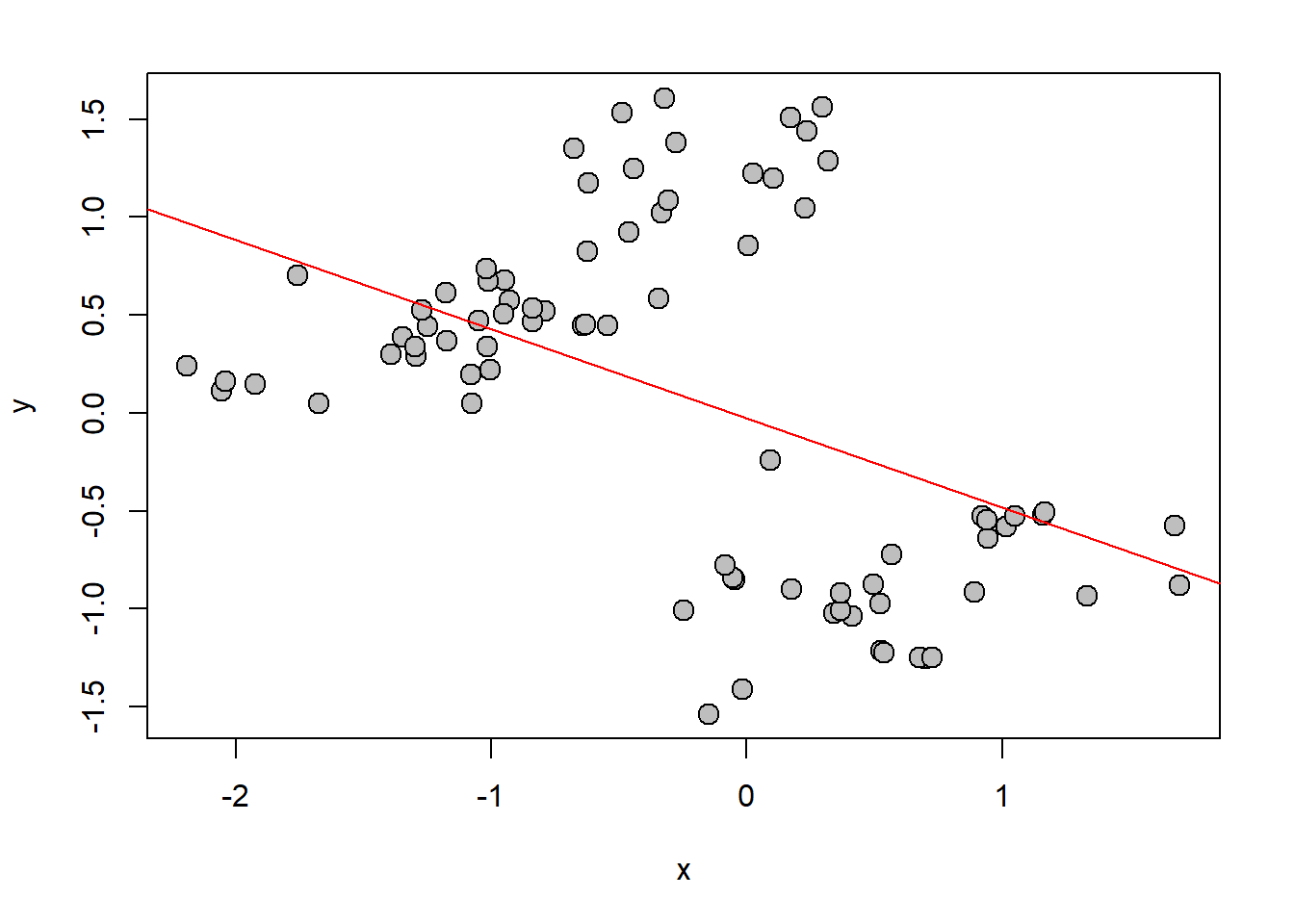
summary(fit.OLS)Mesmo os dados tendo sidos simulados independentemente (na ausência de correlação entre x e y) nós podemos ver que existe uma correlação entre os dados induzida pela filogenia (erro tipo I). Podemos plotar a filogenia no espaço de forma para descobrir o motivo:
{phylomorphospace(tree,cbind(x,y),label="off",node.size=c(0,0))
points(x,y,pch=21,bg="grey",cex=1.5)
abline(fit.OLS,col="red",lwd=2)}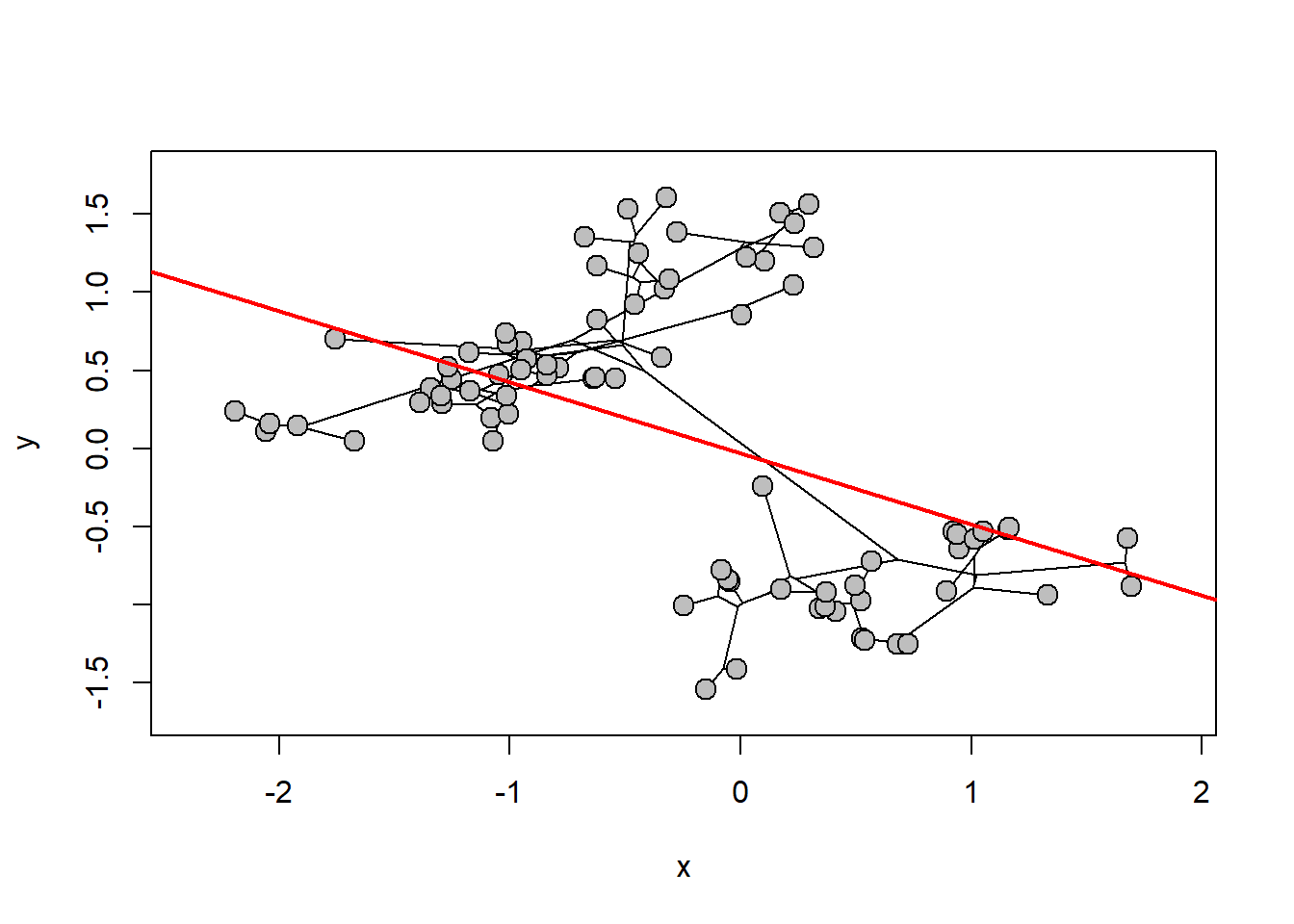 Neste caso, espécies próximas filogeneticamente têm valores de atributo muito similares. Isso gera dois grupos de espécies próximas com fenótipo similar. Podemos usar os contrastes independentes neste caso.
Neste caso, espécies próximas filogeneticamente têm valores de atributo muito similares. Isso gera dois grupos de espécies próximas com fenótipo similar. Podemos usar os contrastes independentes neste caso.
# Ajustando PIC
pic.x<-pic(x,tree)
pic.y<-pic(y,tree)
fit.pic<-lm(pic.y~pic.x+0)
summary(fit.pic)
summary.aov(fit.pic)23.2 Regressão filogenética generalisada por quadrados mínimos
Vários pacotes realizam PGLS no R. Vamos usar os mais comuns - ape/nlme + caper. A PGLS usa uma matriz de covariância entre espécies de acordo com algum processo evolutivo (BM, Pagel, OU, outros).
Vamos carregar alguns dados de borboletas.
require(ape)
dados<-read.table("dadospcm/butterfly-data.txt",h=T,row.names = 1)
dados
attach(dados)
wing_length<-setNames(wing_length,rownames(dados))
temp<-setNames(temp,rownames(dados))
eye_width<-setNames(eye_width,rownames(dados))
# Carregar árvore
tree<-read.tree("dadospcm/butterfly-tree.txt")
{plot(tree)
axisPhylo()}
Conferir correspondência entre espécies nos dados fenotópicos e na árvore filogenética.
require(geiger)
#> Le chargement a nécessité le package : geiger
obj<-name.check(tree,dados)
obj
# 59 espécies na árvore não contêm dados
# remover espécies da árvore
tree.pruned<-drop.tip(tree,obj$tree_not_data)
name.check(tree.pruned,dados)
plot(tree.pruned)
PGLS com os pacotes ape e nlme.
require(ape)
require(nlme)
#> Le chargement a nécessité le package : nlme
# Estrutura de covariação Browniana
BM<-corBrownian(1,tree.pruned)
BM
# Podemos ajustar um modelo PGLS para investigar a relação entre temperatura e
#comprimento da asa
modelo1<-gls(wing_length~temp,data=dados,correlation=BM)
summary(modelo1)
# Plots de diagnóstico
qqnorm(modelo1$residuals)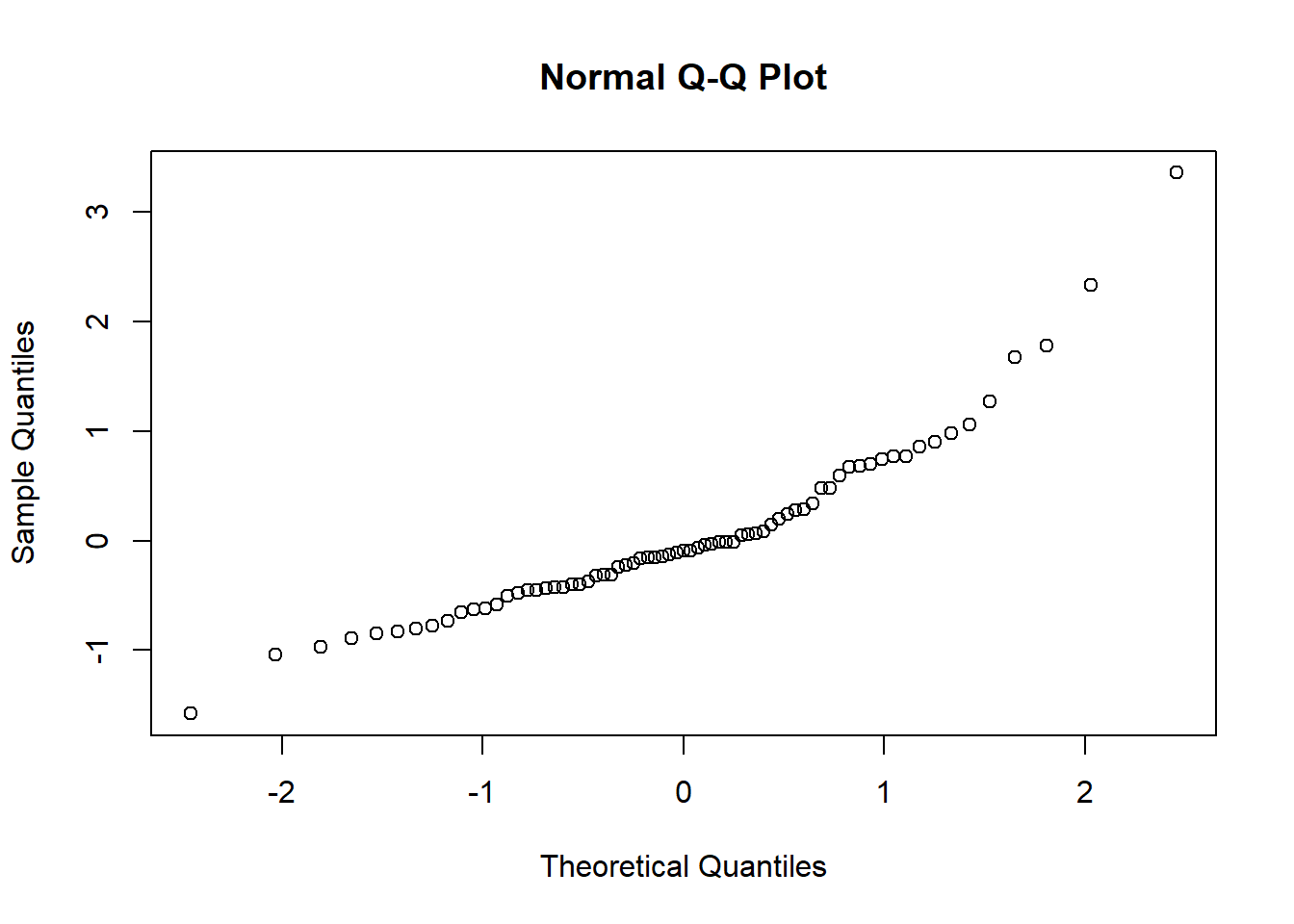
# Comparando com OLS, os resultados são similares
modelo.ols<-lm(wing_length~temp)
summary(modelo.ols)
# Plotando os resultados
{plot(temp,wing_length,pch=21,bg="grey",cex=1.5)
abline(modelo.ols,col="red",lty="dashed",lwd=2)
abline(modelo1,col="blue",lty="dashed",lwd=2)}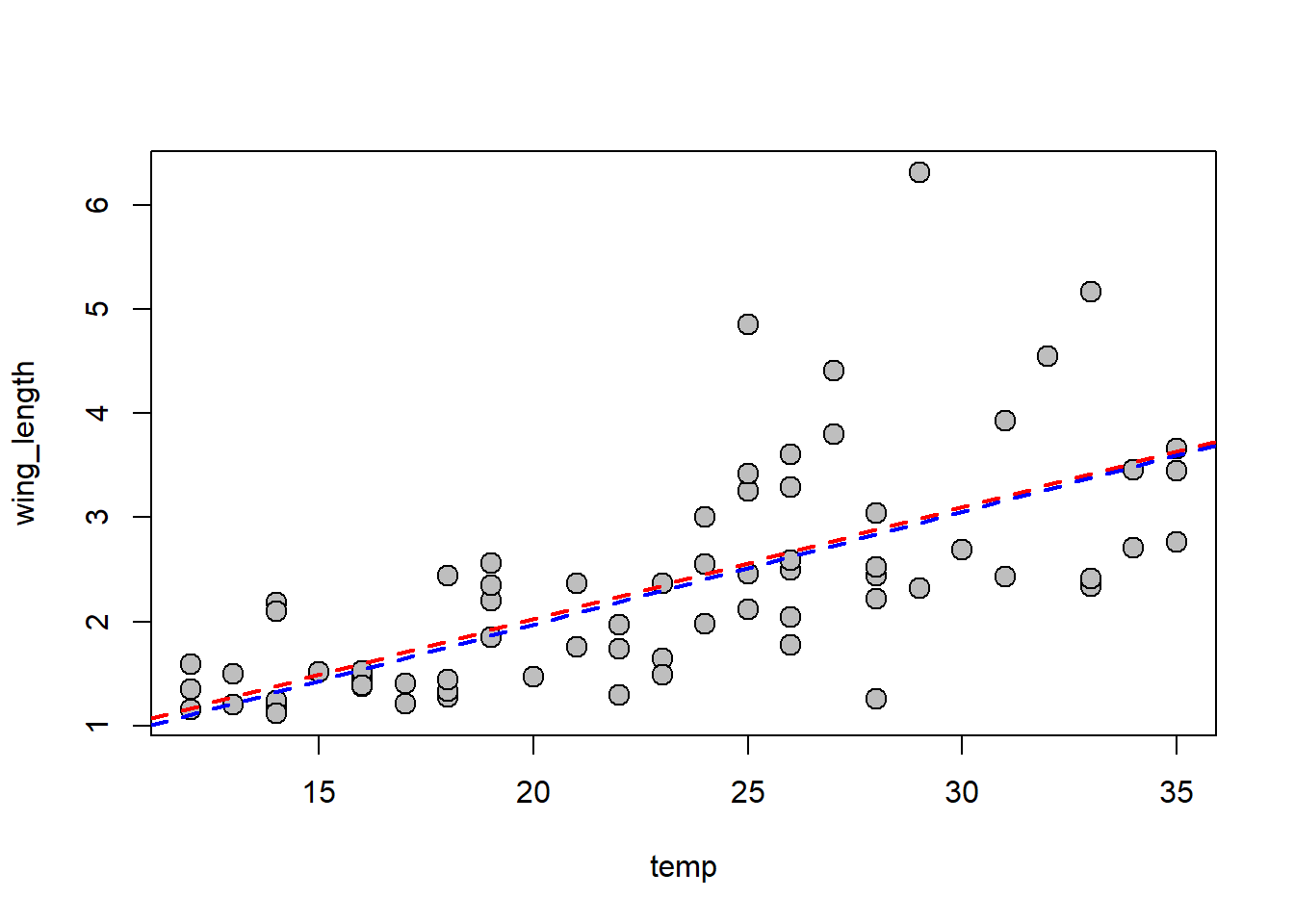
É possível usar outras estruturas de covariância. Um dos modelos mais comuns usa o λ de Pagel. Esse modelo tem um parâmetro adicional (λ) que estima o sinal filogenético nos resíduos; os elementos off-diagonal são multiplicados pelo valor de lambda estimado, que varia entre 0 e 1. Quando λ=0 a covariância entre espécies é zero, e a regressão filogenética = regressão comum. Quando λ=1 a covariação é igual ao esperado pelo modelo Browniano (= PIC).
modelo2<-gls(wing_length~temp,data=dados,correlation=corPagel(1,tree.pruned))
summary(modelo2)
{plot(temp,wing_length,pch=21,bg="grey",cex=1.5)
abline(modelo.ols,col="red",lty="dashed",lwd=2)
abline(modelo1,col="blue",lty="dashed",lwd=2)
abline(modelo2,col="green",lty="dashed",lwd=2)}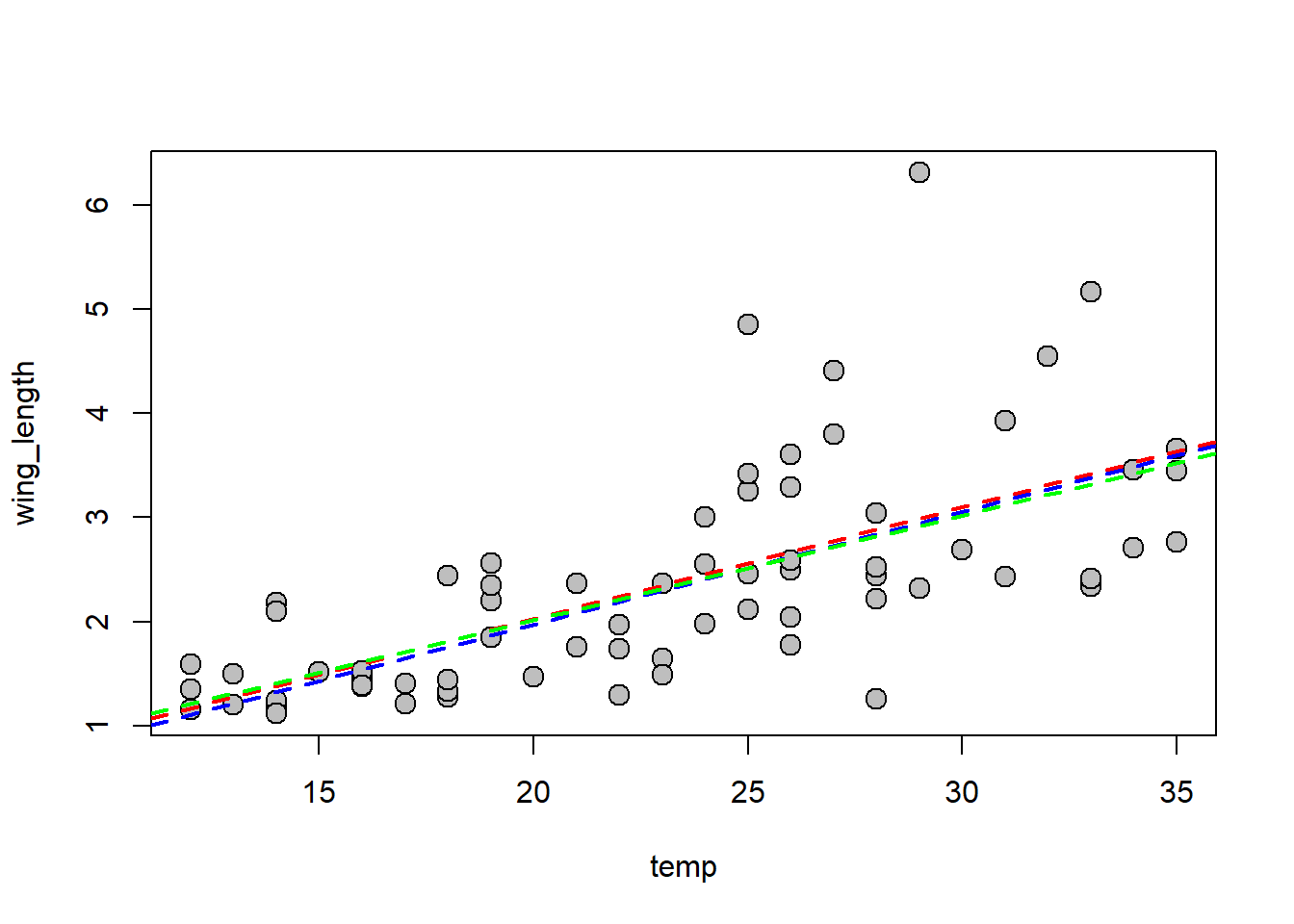
PGLS pode ser aplicada em um contexto de regressão múltipla.
modelo3<-
gls(wing_length~eye_width+temp,data=dados,correlation=corPagel(1,tree.pruned))
summary(modelo3)PGLS com o pacote caper. O caper tem um objeto especial ‘comparative data’ para combinar todos os dados.
require(caper)
#> Le chargement a nécessité le package : caper
#> Le chargement a nécessité le package : MASS
#> Le chargement a nécessité le package : mvtnorm
dados<-read.table("dadospcm/butterfly-data.txt",header=T)
comp.data<-comparative.data(tree.pruned,dados,names.col='species')
# pgls usando lambda
modelo4<-pgls(wing_length~temp,data = comp.data,lambda="ML")
summary(modelo4)Com o caper é possível plotar gráficos de diagnostico facilmente.
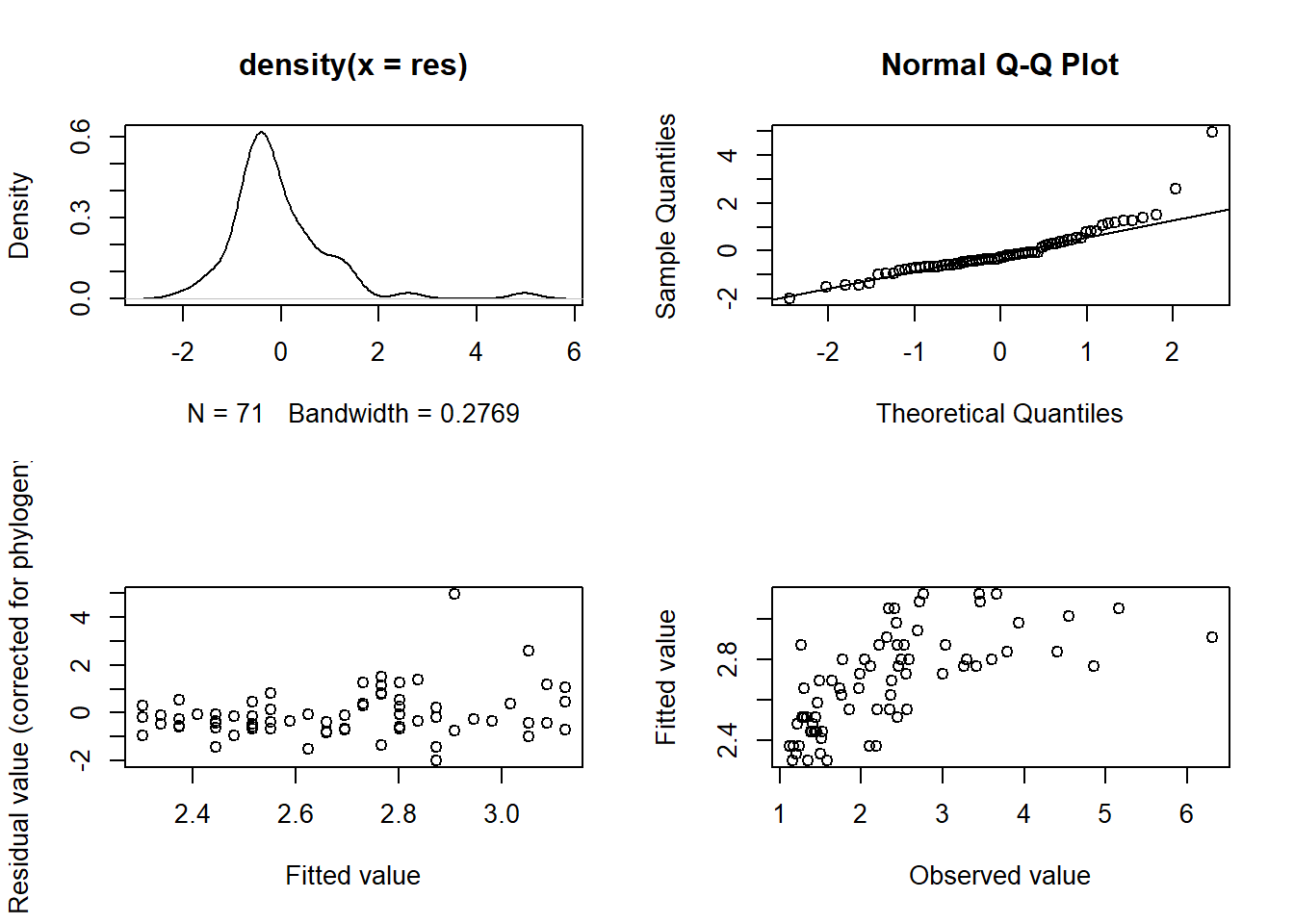
O que olhar nesses plots: No gráfico 1, dados com resíduos maiores que +- 3 podem ser outliers. Os pontos no QQ-plot (gráfico 2) devem cair aproximadamente sobre a linha. Os gráficos 3 e 4 devem mostrar uma distribuição mais ou menos randômica, sem padrões aparentes.
Como qualquer outro modelo linear, também é preciso prestar atenção ao número de preditores e número de termos estimados no modelo em relação ao número deobservações (espécies). Uma “regra” simples é usar +- 10 vezes mais espécies do que termos estimados (incluindo intercepto, interações, e o parâmetro lambda, por exemplo).
23.3 Regressão de autovetores filogenéticos
Regressão de autovetores filogenéticos e Curva PSR com o pacote PVR. Veja o capítulo sobre Sinal Filogenético pra mais detalhes sobre as análises.
require(PVR)
#> Le chargement a nécessité le package : PVR
#> Warning: le package 'PVR' a été compilé avec la version R
#> 4.3.2
#> PVR 0.3 loaded
# Decomposição de matriz de distância filogenética
pvr_obj<-PVRdecomp(tree.pruned)
# Regressão de autovetores filogenéticos
pvr_reg<-PVR(pvr_obj,tree.pruned,wing_length,temp)
# Porcentagem de cada componente
pvr_reg@VarPart
#> $a
#> [1] 0.3326349
#>
#> $b
#> [1] 0.1093753
#>
#> $c
#> [1] 0.08198286
#>
#> $d
#> [1] 0.4760069
VarPartplot(pvr_reg)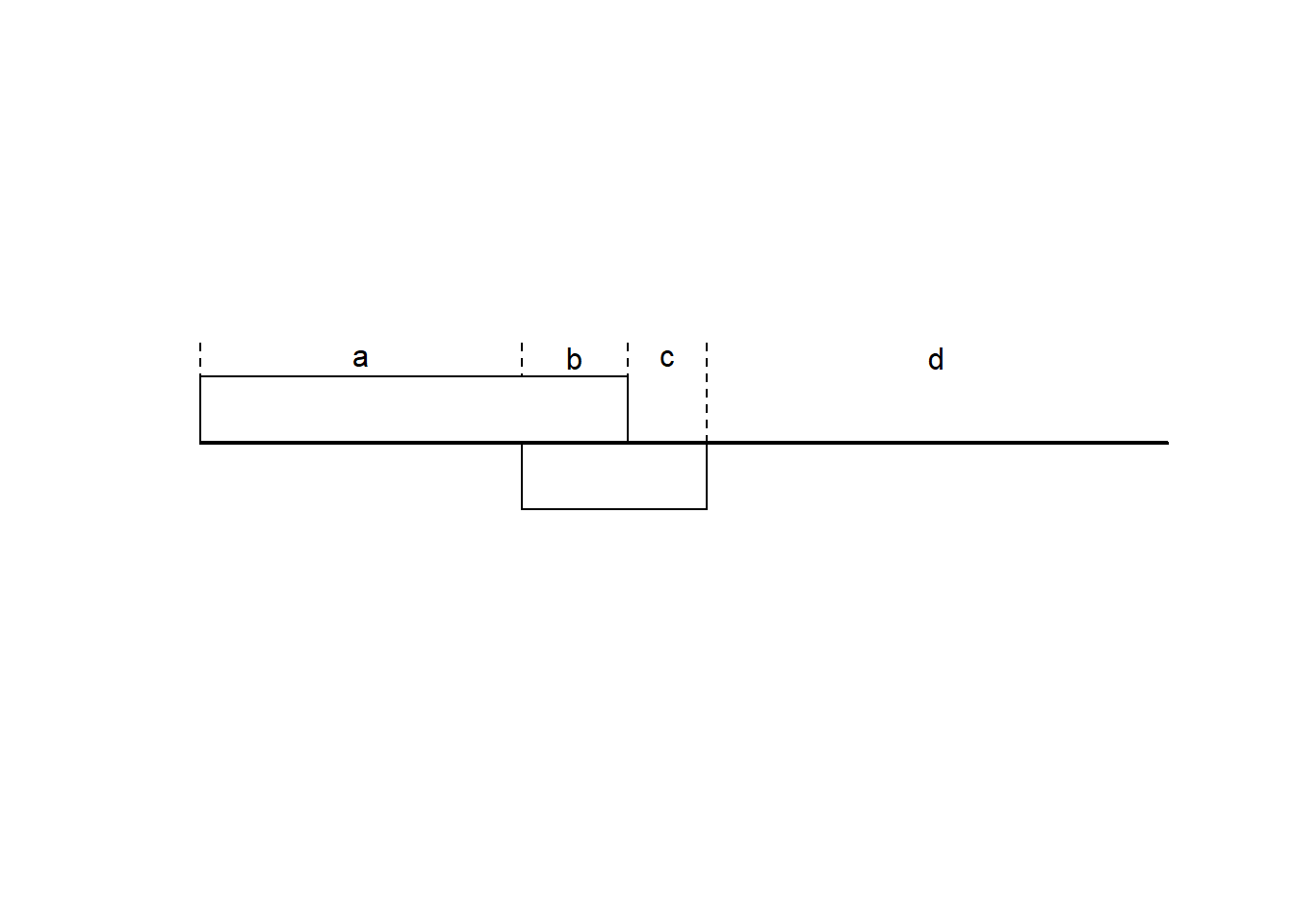
#> [a] - Variation explained by environmental variables
#> [b] - Shared variation between environmental variables and phylogeny(PVR)
#> [c] - Variation explained by phylogeny(PVR)
#> [d] - Unexplained variation
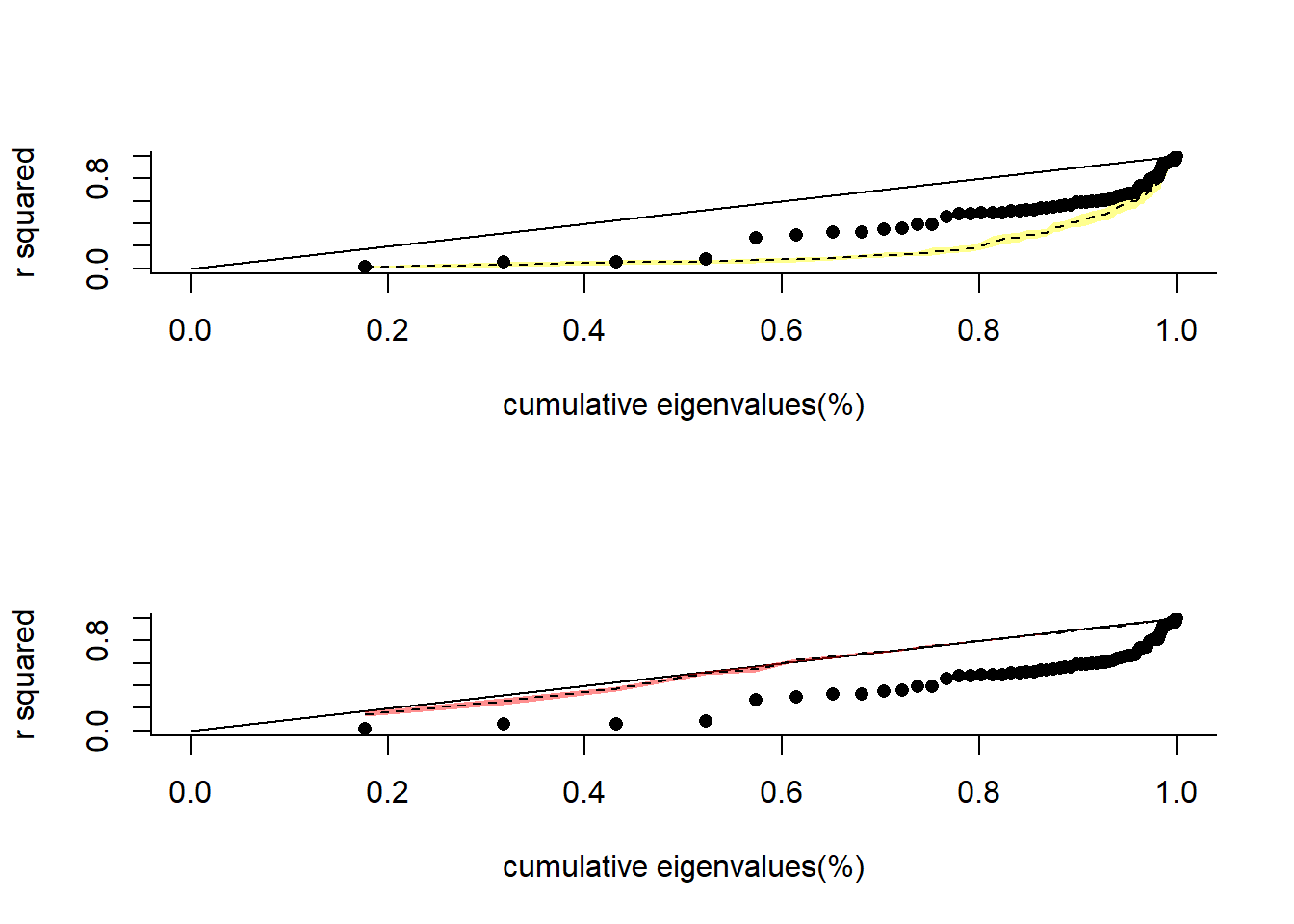
# Sinal Filogenético com PSR Curve
psr_obj<-PSR(pvr_obj,trait=wing_length,
null.model=TRUE,Brownian.model=TRUE,times=10)
PSRplot(psr_obj,info="both")