Capítulo 30 Diversificação dependente de estado
Esse capítulo exemplifica a lógica dos modelos de diversificação dependentes de estado (SSE) usando o modelo mais simples e primeiro a ser desenvolvido, o BiSSE. Há uma grande literatura sobre modelos dependentes de estado, em constante atualização, que precisa ser consultada antes de utilizar um modelo SSE. Veja os artigos nas referências recomendadas na disciplina.
Carregar dados.
require(ape)
#> Le chargement a nécessité le package : ape
require(phytools)
#> Le chargement a nécessité le package : phytools
#> Le chargement a nécessité le package : maps
require(geiger)
#> Le chargement a nécessité le package : geiger
require(diversitree)
#> Le chargement a nécessité le package : diversitree
# Filogenia
tree<-read.tree("dadospcm/primate-tree.txt")
plotTree(tree,fsize=0.4,ftype="i",type="fan",lwd=1)
# Atributos
dados<-read.table("dadospcm/primate-data.txt",h=T,row.names=1)
head(dados)
# Recortar filogenia
match.species<-treedata(tree,dados)
tree<-match.species$phy
plotTree(tree,fsize=0.5,ftype="i",type="fan",lwd=1)
Vamos utilizar o BiSSE (binary-state speciation and extinction) para testar hipóteses sobre a diversificação de primatas e o tamanho do grupo social. Considere a variável binária, que possui dois estados: Solitary vs. Social. Menos de 4 ind = solitary Mais de 4 ind = social
binsocial<-dados$BinSocial
names(binsocial)=rownames(dados)
binsocial<-as.factor(binsocial)
binsocialUma dessas categorias promove maior diversificação em primatas?
# O diversitree requer que os códigos sejam numéricos.
levels(binsocial) # 1-Social, 2-Solitário
binsocial<-as.numeric(binsocial)-1 # valores tem que ser 0 e 1
binsocial
names(binsocial)=rownames(dados)
binsocial # 0-Social, 1-SolitárioO BiSSE tem uma função para estimar valores iniciais ‘razoáveis’ dos parâmetros de interesse.
p<-starting.point.bisse(tree)
pPara rodar o BiSSE temos que criar a função de verossimilhança (likelihood) para posteriormente otimizar os parâmetros.
lik.bisse<-make.bisse(tree,binsocial)
lik.bisseAgora podemos restringir a função geral para criar diferentes modelos. Por exemplo, podemos criar um modelo “nulo” onde as taxas de especiação e extinção sejam iguais entre estados (b0b1;d0d1) e que existe uma única taxa de transição (q01=q10).
lik.null<-constrain(lik.bisse,lambda0~lambda1,mu0~mu1,q10~q01)
lik.nullAjustar os modelos.
# Ajustar modelo nulo
p[argnames(lik.null)] # ajustar número de parâmetros inicial
fit.null<-find.mle(lik.null,x.init=p[argnames(lik.null)])
fit.null
# Ajustar o modelo mais complexo, onde as taxas de especiação e extinção dependem do estado do fenótipo
fit.full<-find.mle(lik.bisse,x.init=p)
fit.fullOutros modelos podem ser ajustados entre o mais simples e o mais complexo, dependendo das hipóteses biológicas a priori. Em um estudo empírico, especialmente com múltiplos estados (MuSSE e similares), os modelos devem ser identificados a priori ao invés de ajustar todos os modelos possíveis para identificar aqueles com melhor ajuste. Como exemplo, vamos ajustar um modelo onde somente a taxa de especiação varie ente estados, um modelo onde somente a taxa de extinção varie, e um modelo com b e d constantes, mas com taxas de transição diferentes entre estados.
# especiação variável
lik.lambda<-constrain(lik.bisse,mu0~mu1,q10~q01)
lik.lambda
# ajuste
fit.lambda<-find.mle(lik.lambda,x.init=p[argnames(lik.lambda)])
fit.lambda
# extinção variável
lik.mu<-constrain(lik.bisse,lambda0~lambda1,q10~q01)
lik.mu
# ajuste
fit.mu<-find.mle(lik.mu,x.init=p[argnames(lik.mu)])
fit.mu
# especiação e extinção variável, única taxa de transição entre estados
lik.lambda.mu<-constrain(lik.bisse,q10~q01)
lik.lambda.mu
# ajuste
fit.lambda.mu<-find.mle(lik.lambda.mu,x.init=p[argnames(lik.lambda.mu)])
fit.lambda.mu
# b e d constantes, taxas de transição flexíveis
lik.q<-constrain(lik.bisse,lambda0~lambda1,mu0~mu1)
lik.q
# ajuste
fit.q<-find.mle(lik.q,x.init=p[argnames(lik.q)])
fit.qAgora podemos comparar os modelos ajustados. A função genérica anova permite obter os valores de AIC para uma lista.
# Comparando os modelos
resultados<-anova(fit.null,
completo=fit.full,
lambda.variavel=fit.lambda,
mu.variavel=fit.mu,
lambda.mu.variavel=fit.lambda.mu,
q.variavel=fit.q)
resultados
aicw(setNames(resultados$AIC,rownames(resultados)))
aic.w(setNames(resultados$AIC,rownames(resultados)))Os modelos com especiação variável e com especiação e extinção variável (e taxa de transição única) receberam maior suporte.
coef(fit.lambda.mu)Parece que tanto as taxas de especiação como de extinção são maiores para o estado 0 (social) do que para o estado 1 (solitário). E a taxa de diversificação?
lambda0<-coef(fit.lambda.mu)[[1]]
mu0<-coef(fit.lambda.mu)[[3]]
r.0<-lambda0-mu0
r.0
lambda1<-coef(fit.lambda.mu)[[2]]
mu1<-coef(fit.lambda.mu)[[4]]
r.1<-lambda1-mu1
r.1Como as taxas de extinção estimadas são muito baixas, as taxas de diversificação são próximas das de especiação.
Assim como vimos no modelo bd simples, com o pacote diversitree podemos rodar análises bayesianas com os nossos modelos. Vamos fazer isso para o nosso melhor modelo para estimar os valores dos parâmetros.
# MCMC
mcmc.lambda.mu<-mcmc(lik.lambda.mu,fit.lambda.mu$par, nsteps = 1000, w = 1,print.every=100)
# visualizar probabilidades
colors<-setNames(c("red","blue"),1:2)
par(mfrow=c(1,1),mar=c(5,4,2,2))
# especiação
profiles.plot(mcmc.lambda.mu[,grep("lambda",colnames(mcmc.lambda.mu))],
col.line=colors, las=1, legend.pos="topright")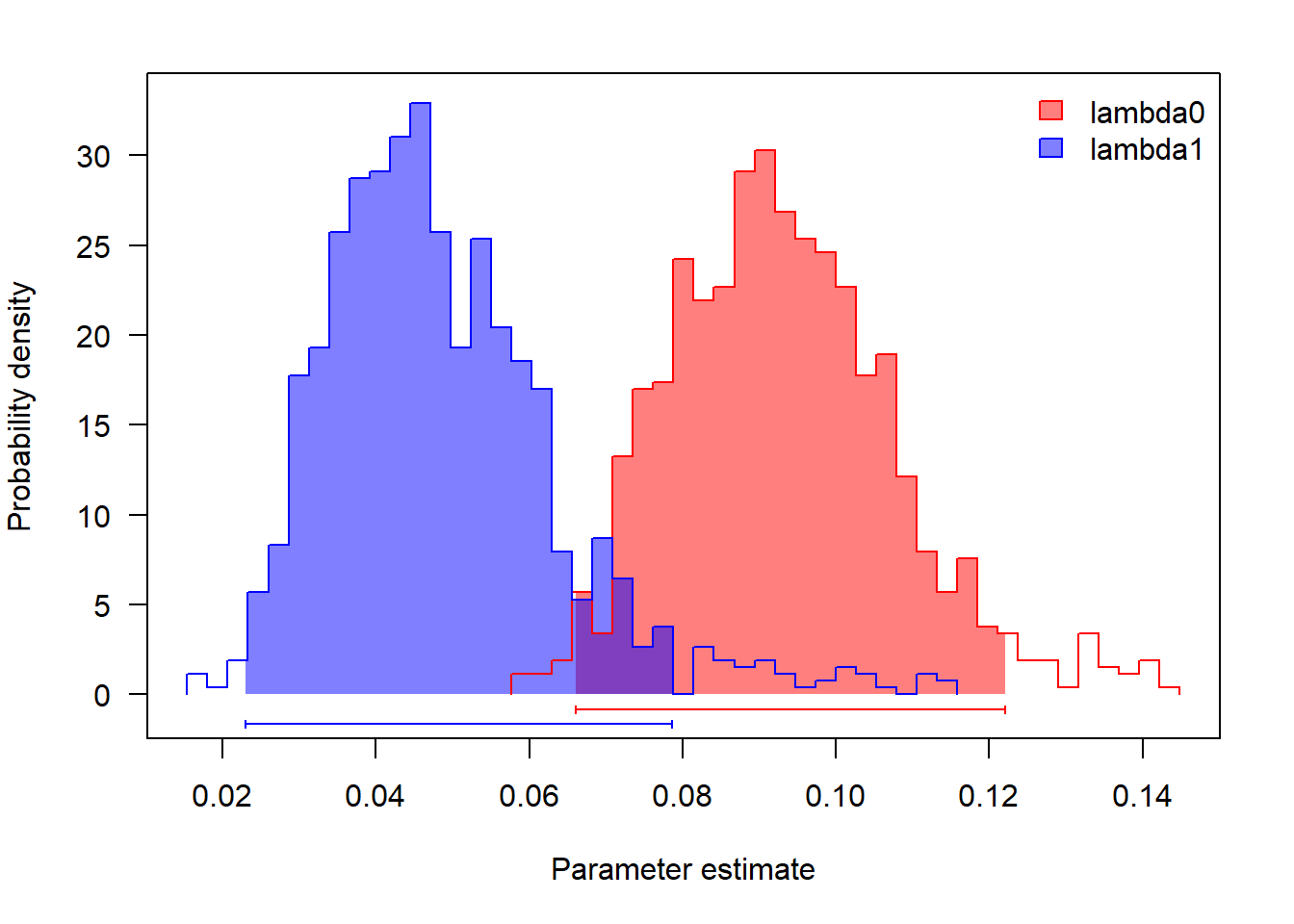
# extinção
profiles.plot(mcmc.lambda.mu[,grep("mu",colnames(mcmc.lambda.mu))],
col.line=colors, las=1, legend.pos="topright")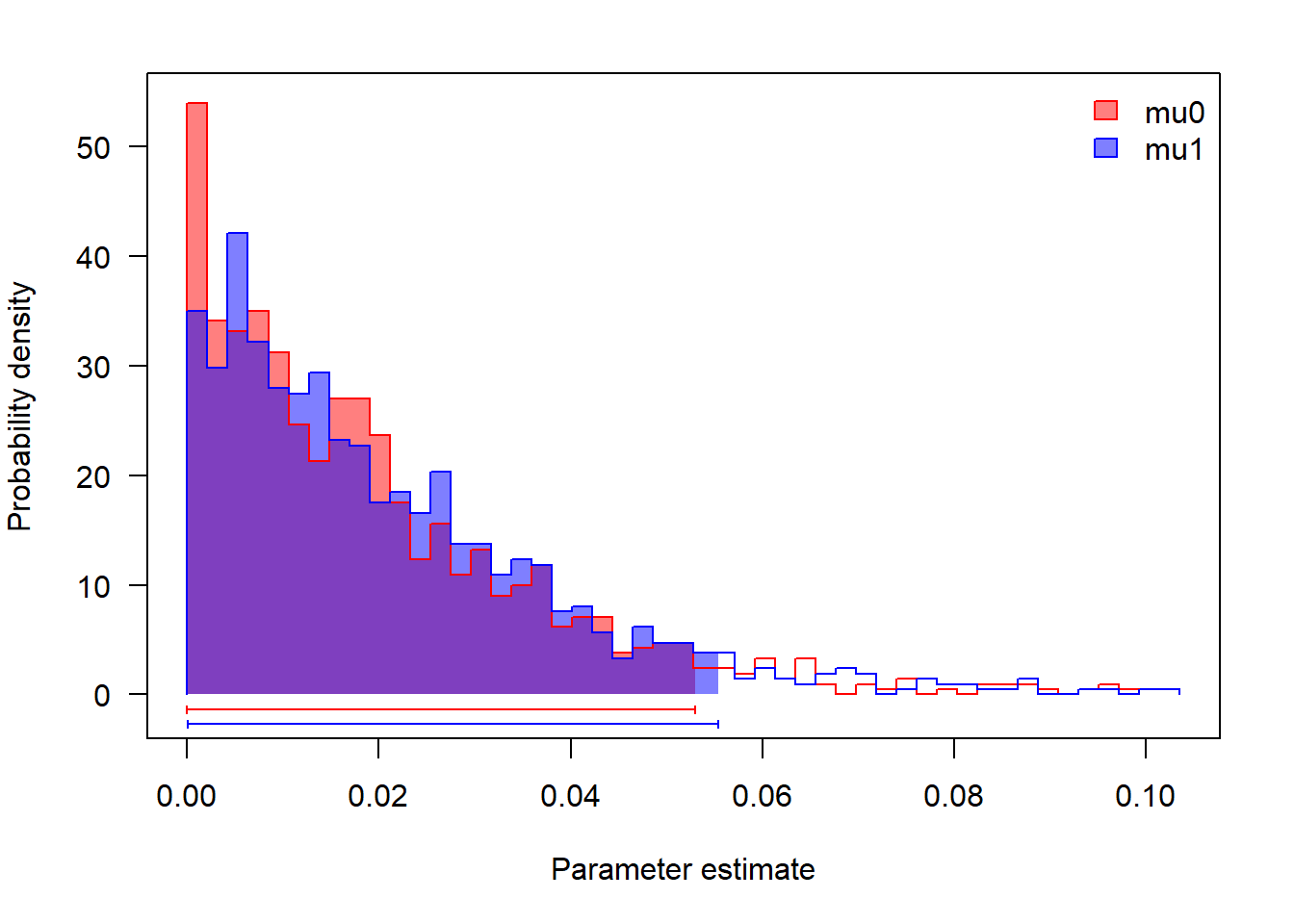
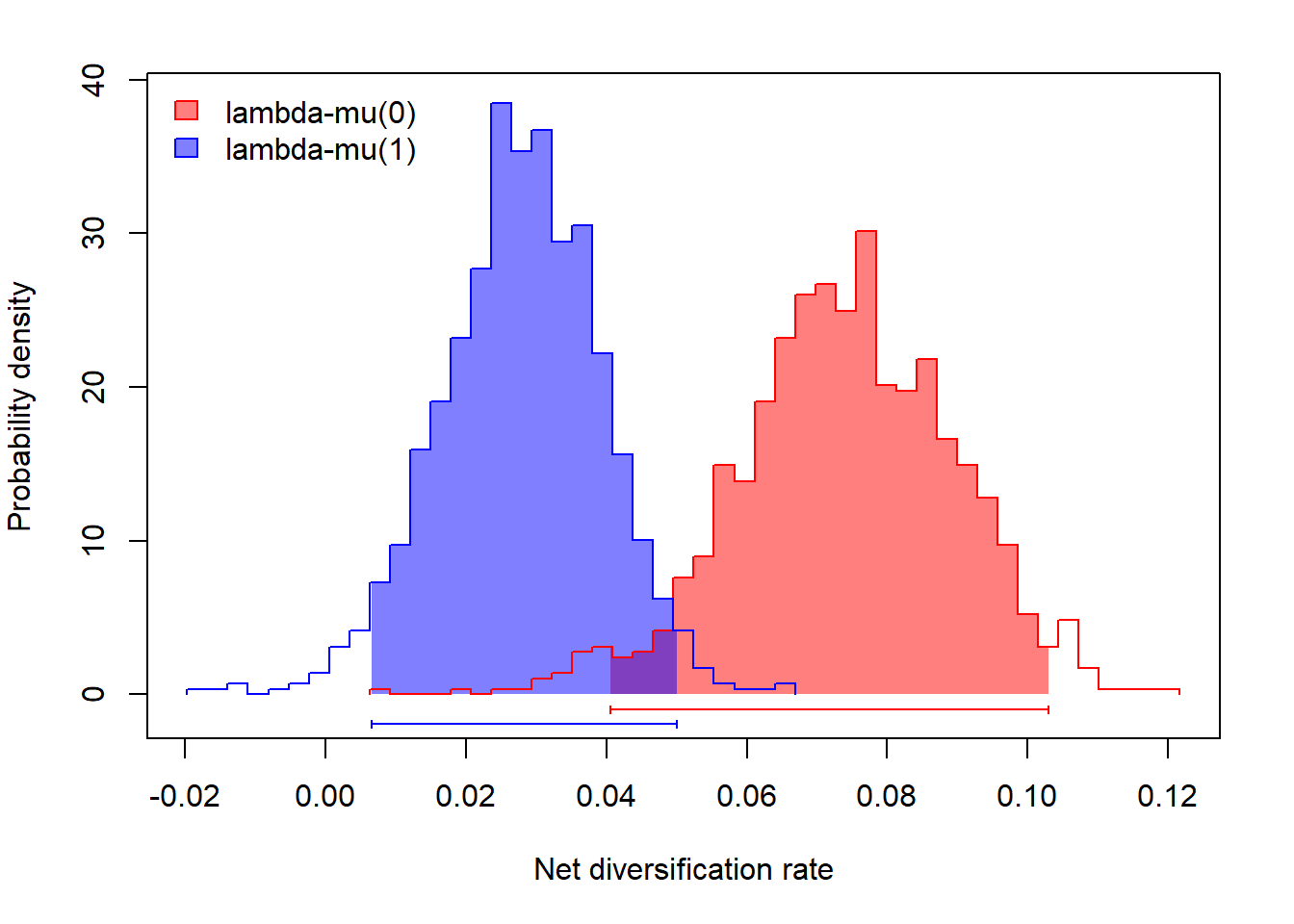
# diversificação líquida
div<-mcmc.lambda.mu[,grep("lambda",colnames(mcmc.lambda.mu))]-
mcmc.lambda.mu[,grep("mu",colnames(mcmc.lambda.mu))]
colnames(div)<-paste("lambda-mu(",0:1,")",sep="")
profiles.plot(div,
xlab="Net diversification rate", ylab="Probability density",
legend.pos="topleft",col.line=setNames(colors,colnames(div)),
lty=1)