Capítulo 32 Co-diversificação
Análises cofilogenéticas investigam a associação/dependência entre a diversificação de dois clados, normalmente um clado de parasitas e um clado de hospedeiros; para uma revisão veja Perez-Lamarque and Morlon (2022). Aqui, usaremos um teste baseado em Mantel para testar o sinal filogenético na associação entre dois clados (Perez-Lamarque and Morlon 2022). Vamos carregar dados de associação entre coronavirus (parasitas) e mamíferos (hospedeiros) de Maestri et al. (n.d.).
Carregar dados.
# Carregar árvore de Coronaviridae
require(ape)
#> Le chargement a nécessité le package : ape
cov_tree<-read.nexus("dadospcm/combined_alignment_cov_trimal_tree_35.tre")
# Carregar matriz de associação
hosts_matrix<-read.table("dadospcm/association_matrix.txt")
# Carregar árvore de mamíferos hospedeiros
hostmammaltree<-read.tree("dadospcm/hostsMammalTree.tre")Sinal filogenético na associação, com permutações mantendo constante o número de parceiros.
require(RPANDA)
#> Le chargement a nécessité le package : RPANDA
#> Le chargement a nécessité le package : picante
#> Le chargement a nécessité le package : vegan
#> Le chargement a nécessité le package : permute
#> Le chargement a nécessité le package : lattice
#> This is vegan 2.6-4
#> Le chargement a nécessité le package : nlme
psuni_n<-phylosignal_network(hosts_matrix, tree_A = cov_tree,
tree_B = hostmammaltree,
method = "GUniFrac",
correlation = "Pearson",
nperm=1000, permutation = "nbpartners")
psuni_nCoronavirus próximos filogenéticamente tendem a se associar (infectar) mamíferos proximos filogenéticamente (r=0.377) e vice-versa (r=0.295).
Também podemos testar se parasitas proximamente relacionados tendem a infectar um número de hospeiros similar.
# Sinal filogenético no número de parceiros
npart_cov<-phylosignal_network(hosts_matrix,
tree_A = cov_tree,
method = "degree",
correlation = "Pearson", nperm = 1000)
npart_mammals<-phylosignal_network(t(hosts_matrix),
tree_A = hostmammaltree,
method = "degree",
correlation = "Pearson", nperm = 1000)
npart_cov
npart_mammalsCoronavirus próximos filogenéticamente tendem a infectar um número de hospeiros similar, porém, mamíferos proximos filogenéticamente não tendem a hospedar um número similar de coronavirus. Isso sugere que a especificidade de coronavírus em relação aos seus hospedeiros é conservada evolutivamente, enquanto que a suscetibilidade dos hospedeiros aos coronavírus não é.
Por fim, podemos testar em que escala filogenética o sinal é mais forte.
# Calcular sinal filogenético por subclados
# Para parasitas
results_clade_A <- phylosignal_sub_network(hosts_matrix,
tree_A = cov_tree,
tree_B = hostmammaltree,
method = "GUniFrac",
correlation = "Pearson",
nperm=1000,minimum=10, degree=F,
permutation = "shuffle")
plot_phylosignal_sub_network(tree_A = cov_tree, results_clade_A,
network = hosts_matrix)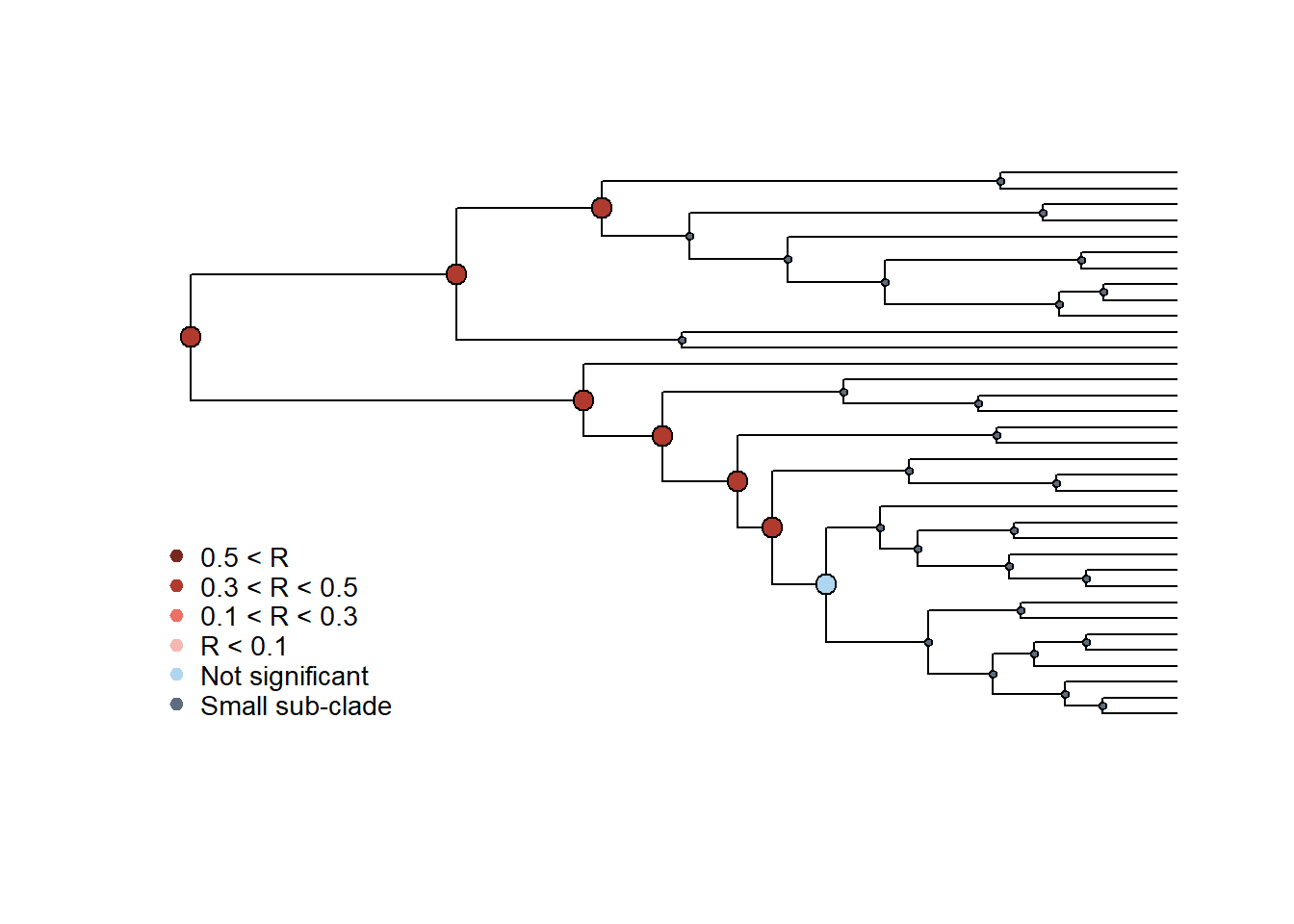
# Para hospedeiros
results_clade_B <- phylosignal_sub_network(t(hosts_matrix),
tree_A = hostmammaltree,
tree_B = cov_tree,
method = "GUniFrac",
correlation = "Pearson",
nperm=1000,
minimum=10, degree=F,
permutation = "shuffle")
plot_phylosignal_sub_network(tree_A = hostmammaltree, results_clade_B,
network = t(hosts_matrix),
legend=FALSE,where = "topleft")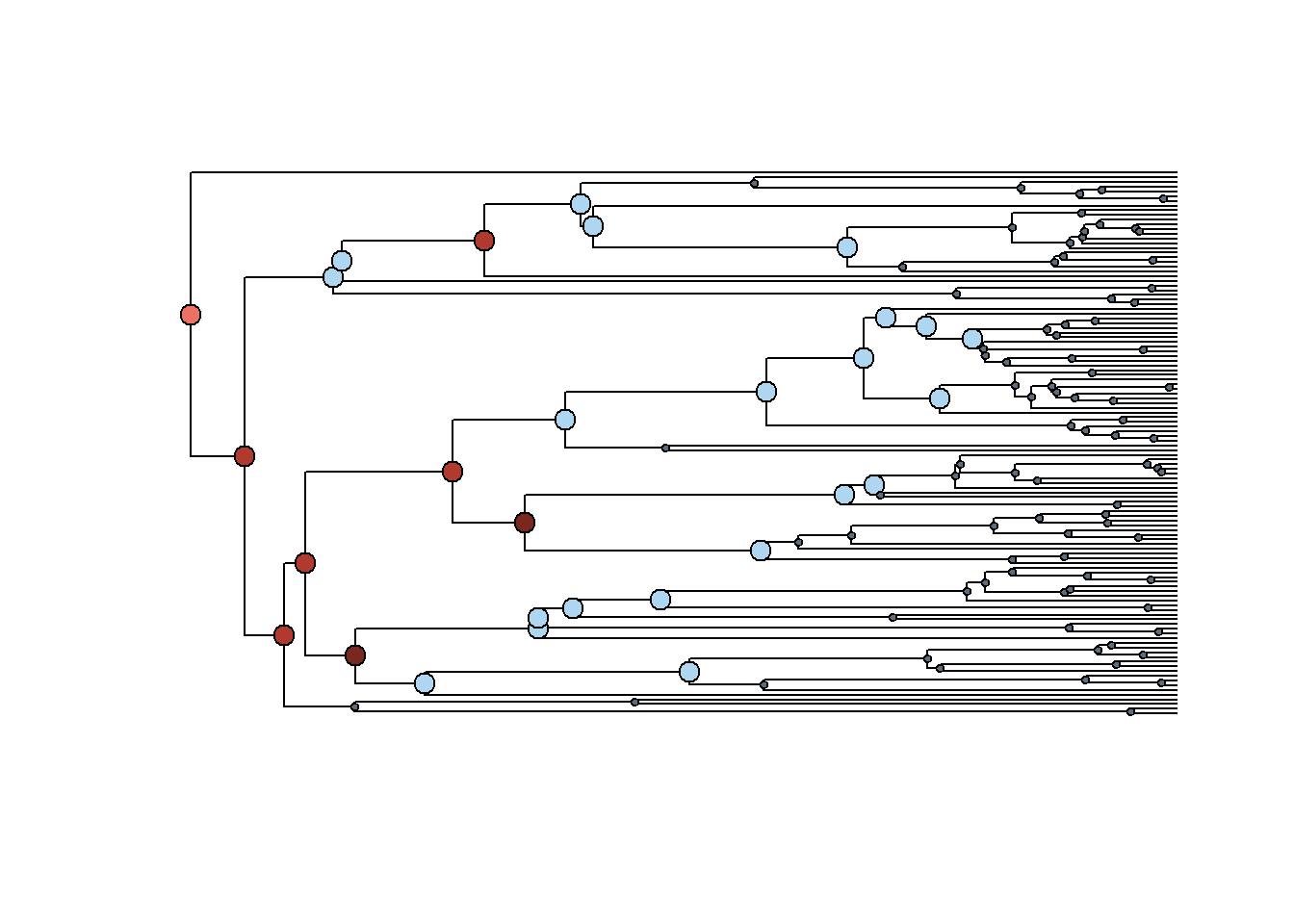
# Plot conjunto
layout(matrix(c(1,2),1,2),widths=c(0.5,0.5),heights=c(0.1,0.1))
plot_phylosignal_sub_network(tree_A = cov_tree, results_clade_A,
network = hosts_matrix)
plot_phylosignal_sub_network(tree_A = hostmammaltree, results_clade_B,
network = t(hosts_matrix),
legend=FALSE,where = "topleft")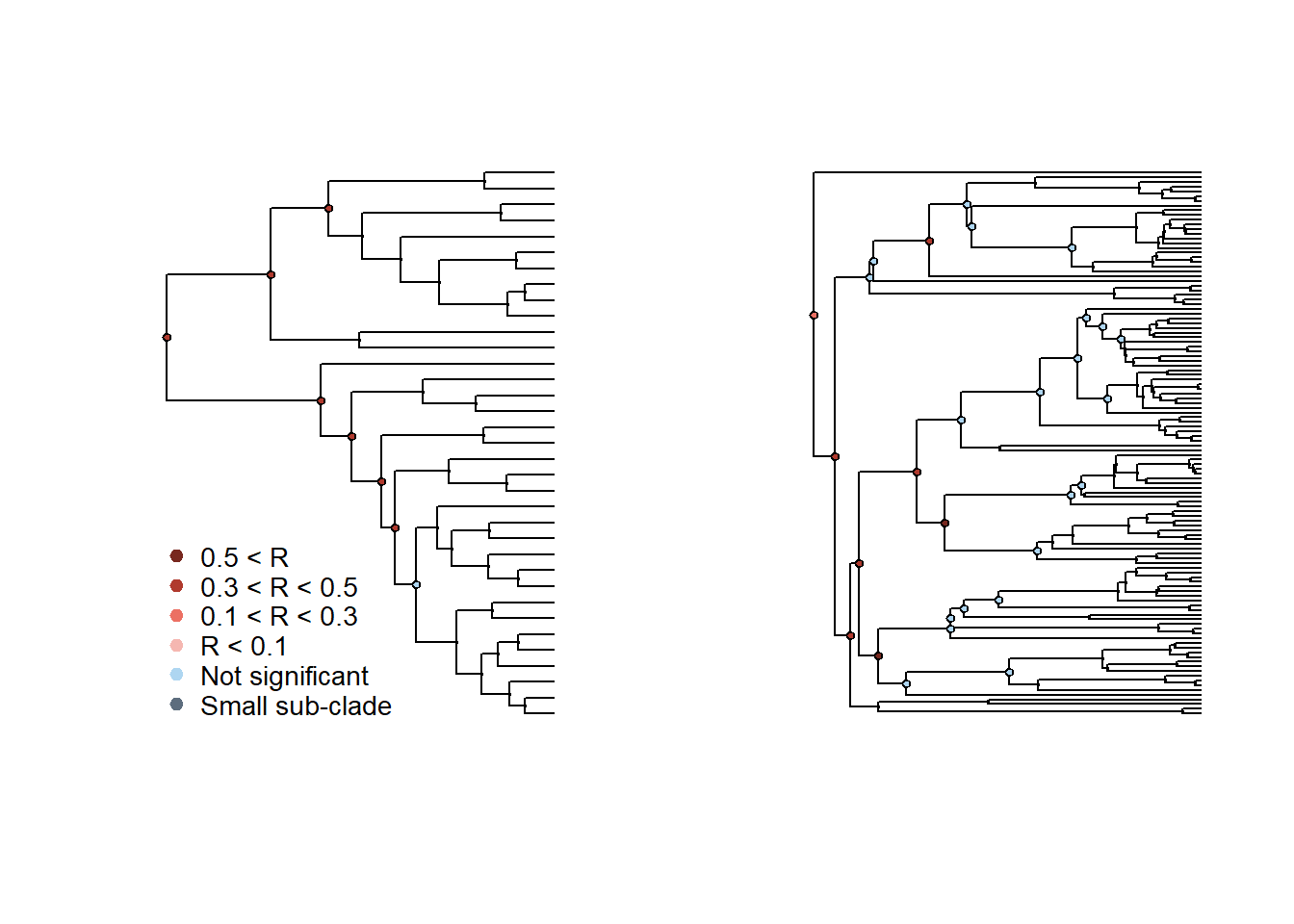
Em geral, o sinal filogenético é mais forte nos nós mais antigos das duas filogenias.